ARTERITIS
2003 werd beschreven. Op dit moment
zijn in de literatuur 15 gevallen gepu-
bliceerd. Het klinische beeld bestaat
pigmenteerde, netvormige, niet-geïn-
filtreerde huidvlekken van de onderste
ledematen. De romp is zelden aangetast.
De letsels zijn asymptomatisch en jeu-
ken niet. De aandoening mag niet wor-
den miskend omdat ze in tegenstelling
tot andere vormen van vasculitis geen
andere organen dan de huid aantast en
gunstig evolueert.
Dit syndroom moet om drie redenen als
een afzonderlijke entiteit worden be-
schouwd:
- een klinisch voorkomen van vasculitis;
- een specifieke histologie met een
fibrinoïde necrose en een intravascu-
laire trombose;
tologie (necroserende arteritis van de
middelgrote bloedvaten).
schreven: corticosteroïden voor de huid,
plaquenil, disulone, aspirine en algemene
corticotherapie met een beperkte doel-
treffendheid. NSAID's zijn ook weinig
doeltreffend. De differentiële diagnose
moet worden gesteld met enerzijds poly-
arteritis nodosa die ook de middelgrote
bloedvaten aantast (fibrinoïde necrose,
maar een polymorfer infiltraat van lym-
focyten en eosinofielen) en anderzijds
livedovasculitis zonder specifiek infiltraat
bij de histologie.
twee specifieke situaties voorkomen:
bulleuze lupus in zijn beginstadium en
in het kader van intense systeemlupus
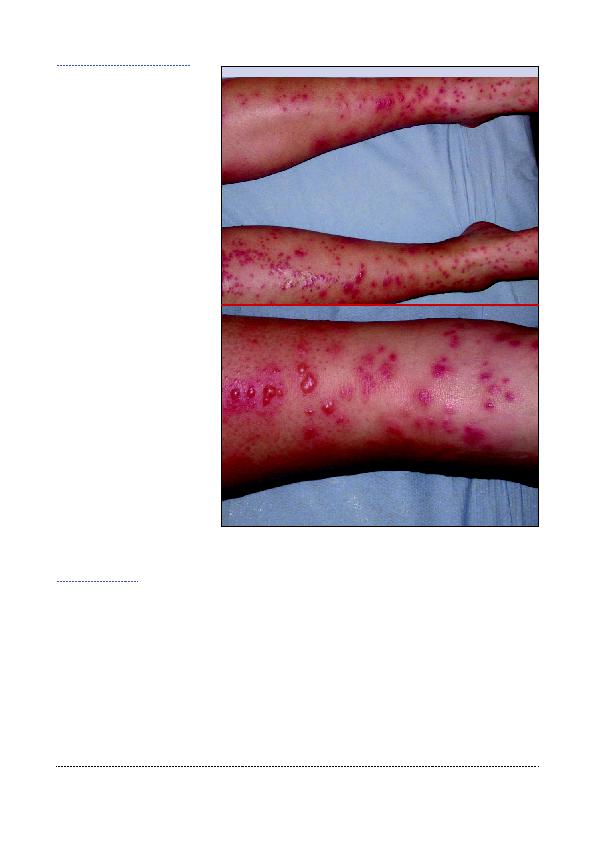
die `blaren krijgt'. Bulleuze lupus in de
beginfase verschijnt abrupt en komt
doorgaans voor bij jonge volwassenen
die mogelijk systemische lupus erythe-
matodes (SLE) hebben. De aandoening
wordt gekenmerkt door een veralge-
meende uitslag met vesikels en blaren
die zowel de blootgestelde als niet-
blootgestelde huid aantast. De vesikels
uitwaaie ren. Dat geeft een klinisch beeld
dat lijkt op dat van herpetiforme derma-
titis. De diagnose kan worden gesteld
met een histologisch onderzoek: aan-
wezigheid van subepidermale blaren,
een beeld van leukocytoclastische vas-
culitis met een infiltraat van neutrofiele
granulocyten. Bij directe immunofluo-
rescentie wordt een lineaire neerslag
van immunoglobulines IgG, IgM en IgA
en complement C1q, C3 en C4 langs
het gebied van de dermo-epidermale
junctie gezien. Immunoblot toont IgG-
antistoffen die gericht zijn tegen col-
lageen VII, zoals bij verworven epider-
molysis bullosa. Bulleuze lupus reageert
corticosteroïden (dapson in een dosis
van 25-50mg/kg/dag).
scheiden van intense lupus `die blaren
krijgt' waarbij de blaren loskomen ten
gevolge van een ontstekingsproces en in-
tense necrose. Ze komen voor in een zone
die aan licht is blootgesteld en gaan altijd
gepaard met een erythemateuze huiduit-
slag met papels, soms morbiliform, die op
een toxische epidermale necrolyse (TEN)
kan lijken. De histologie is ook anders. Er
is geen subepidermale loslating, maar ke-
ratinocytaire degeneratie. Er worden ook
geen antistoffen teruggevonden die ge-
richt zijn tegen collageen type VII.