PANNICULITISACHTIGE
T-CELLYMFOMEN
lymfoom (SPTL) is een zeldzame vorm
van huidlymfoom. De aandoening uit
zich doorgaans in de vorm van erythe-
mateuze, soms geülcereerde plaques of
noduli, bij voorkeur op de bovenste en
onderste ledematen. De klinische en sys-
temische symptomen zijn niet specifiek
en kunnen bestaan uit koorts, rillingen
en gewichtsverlies (B-symptomen); on-
geveer de helft van de patiënten ontwik-
kelt cytopenie. De ernstige vormen gaan
gepaard met hepatosplenomegalie,
muceuze ulceraties, sereuze exsudaten,
hemofagocytosesyndroom en pancyto-
penie.
scheiden, geklasseerd volgens de T-cel-
receptoren (TCR) en de immunofeno-
typische kenmerken.
PANNICULITISACHTIG
T-CELLYMFOOM VAN HET ALFA/
BÈTATYPE ( TCR)
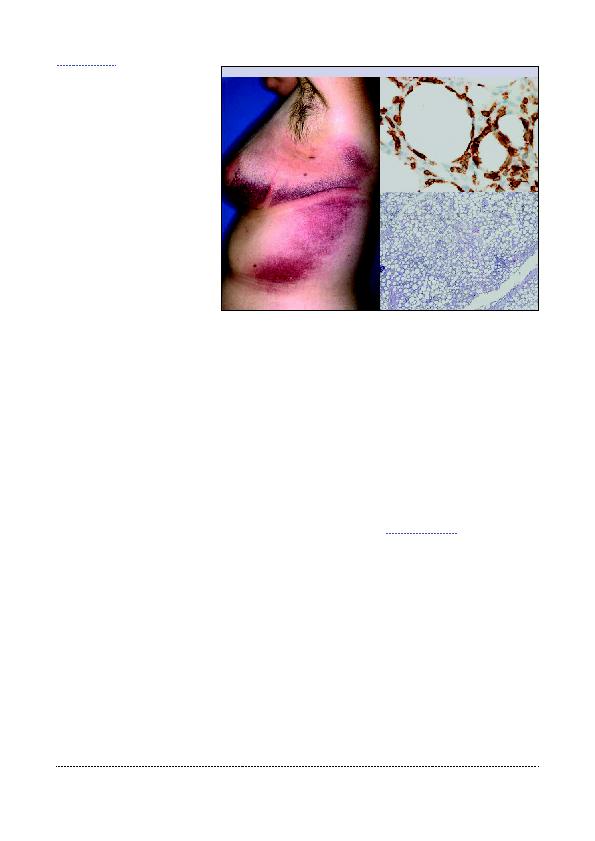
vrouwelijke jonge volwassenen treft.
Het wordt gekenmerkt door subcutane
noduli of min of meer ontstoken plaques
die zich meestal op de onderste lede-
maten bevinden (Figuur 5). Deze vorm
is pijnloos, evolueert langdurig, maar re-
gresseert soms spontaan. In 50% van de
gevallen worden algemene symptomen
vastgesteld. Niet zelden gaat het gepaard
met een hemofagocytosesyndroom.
Daarom moet een beenmergbiopsie ge-
beuren. Uitzaaiing buiten de huid blijft
uitzonderlijk (perifere adenopathieën of
hepatosplenomegalie). De tumorcellen
brengen de cytotoxiciteitmarkers tot ex-
pressie (CD3+, CD8+, CD4-, alfa/bèta+,
CD56-). Het overlevingspercentage is
85% na 5 jaar.
PANNICULITISACHTIG
T-CELLYMFOOM VAN HET GAMMA/
DELTATYPE
klinische achteruitgang en in bijna de helft
van de gevallen met een macrofagenacti-
vatiesyndroom. Andere symptomen dan
doening evolueert abrupt, acuut en de
prognose is gereserveerd. De tumorcel-
len brengen CD4-, CD8- en CD56+ tot
expressie. De diagnose SPTL is gebaseerd
op de klinische kenmerken van de let-
sels, het histopathologische onder-
zoek van de huid- en onderhuidweef-
sels, moleculaire analyse en modellen
voor immunohistochemische kleuring
en immunofenotypering. Een onder-
zoek met medische beeldvorming
sels op andere plaatsen dan de huid op
te sporen om na te gaan hoe verspreid
de aandoening is, maar ook als follow-up
en om de respons op de behandelingen
na te gaan. Een beenmergbiopsie is no-
dig om een eventueel hemofagocytose-
syndroom (HFS) te diagnosticeren, een
belangrijke factor in de behandeling en
prognose van deze lymfomen.
ferentieerd van de andere goedaardige
oorzaken van lobulaire panniculitis, zoals
lupuspanniculitis die klinisch en histolo-
gisch verward kan worden met SPTL. In
beide gevallen wordt immers een beeld
van lymfocytaire panniculitis gezien.
Het is dus belangrijk om te zoeken naar
atypische lymfoïde cellen die zullen wij-
zen op een subcutaan panniculitisachtig
lymfoom. Morfologische argumenten
voor lupus zijn andere histologische te-
kens van lupus zoals een dermatose van
hoorning of neerslag van mucine in de
huid. We merken op dat lupuspanniculitis
niet zelden een subcutaan panniculitis-
achtig T-lymfoom voorafgaat.
behandeling van SPTL's afhangen van de
combinatie met een HFS en een onder-
liggende auto-immuunaandoening (AIA).
Zonder HFS en AIA is de geschikte be-
handeling prednison +/- methotrexaat.
In combinatie met een AIA, zonder HFS
is dat prednison. Als een HFS is aan-
getoond, is de keuzetherapie chemothe-
rapie van het type CHOP.
POLYCHONDRITIS (APC)
zeldzame aandoening die voortvloeit uit
een ontsteking met degeneratie van het
kraakbeen van de oren, neus, larynx en
tracheo-broncheale boom die kan leiden
tot definitieve atrofie. Het kan gepaard
gaan met andere symptomen die het
kraakbeen niet treffen, zoals aantasting van
de huid, gewrichten, ogen, het gehoor, de
bloedvaten, het hart en het bloed. Chon-
dritis is essentieel om APC te kunnen
diagnosticeren. De frequentste eerste
vindplaats is die van aantasting van het
buitenoor, met in het acute stadium een
rode, warme en pijnlijke zwelling die het
(niet-kraakbenige) lelletje ongemoeid laat