pariétales
constituent l'une des rares formes de
polymicrogyries bilatérales dont les
bases moléculaires sont identifiées. Dans
les familles consanguines, elles sont
liées à des mutations dans le gène
GPR56.
gyrie bifronto-pariétale bilatérale ont
également un tableau clinique particu-
lier qui semble spécifique, comportant 5
items cliniques.
On retrouve:
ments oculaires non conjugués (dans
85% des cas), qui prédominent dans
la poursuite oculaire horizontale,
avec une ésotropie et un strabisme,
probablement en rapport avec une
atteinte des noyaux oculomoteurs;
tant avant 5 ans et comportant des
crises généralisées tonico-cloniques;
bien que le pattern EEG soit assez
mal connu, la sévérité de cette épi-
lepsie est en lien avec l'étendue des
polymicrogyries et avec l'implication
de la région frontale;
midaux;
est normal (9).
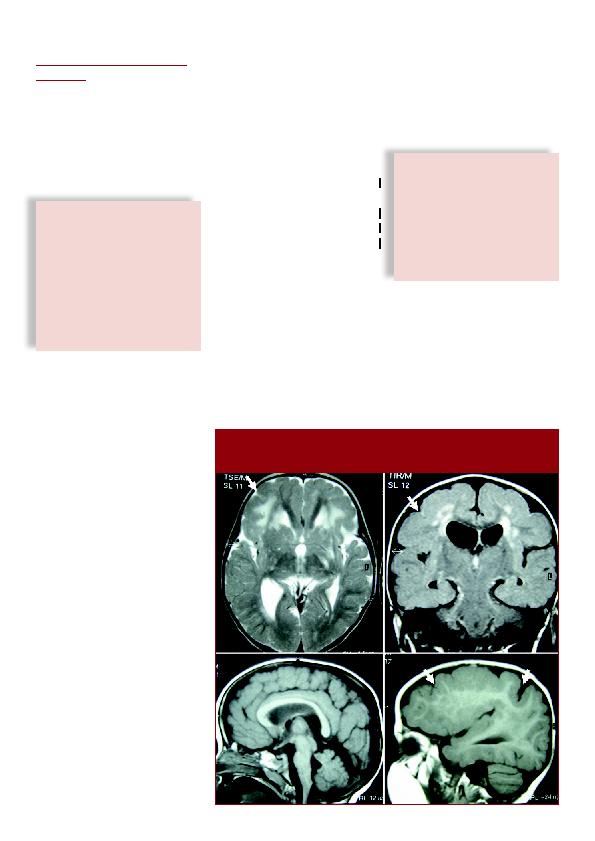
s'étendent de la région frontale à la ré-
gion pariétale, avec un gradient antéro-
postérieur, et incluent la région péri-
rolandique (Figure 3). Dans les formes
liées aux mutations de GPR56, on re-
trouve des hypersignaux en motte de la
substance blanche, témoins de zones de
démyélinisation et une hypoplasie du
tronc cérébral et du cervelet. Les gan-
glions de la base sont normaux.
ont été décrites dans des familles
consanguines, suggérant une hérédité
autosomale récessive, avec des mutations
dans un gène majeur GPR56. Ce gène
code pour un récepteur couplé à une
protéine G, qui semble jouer un rôle
cerveau, et probablement dans la
destinée neuronale (mort neuronale), en
raison de son expression dans les
progénitures neuronales et pas dans les
cellules post-mitotiques.
que la perte de fonction de GPR56 serait
responsable d'un défaut d'intégrité de la
membrane piale, qui constitue une bar-
rière physiologique à la migration des
neurones associée à une anomalie de la
glie radiaire. Ces anomalies entraînent,
chez la souris GPR56 knock-out, une
d'une polymicrogyrie fronto-pariétale et d'une atteinte massive de la substance blanche en
«flaques» ou en motte d'hypersignal T2 (A) ou FLAIR (B).