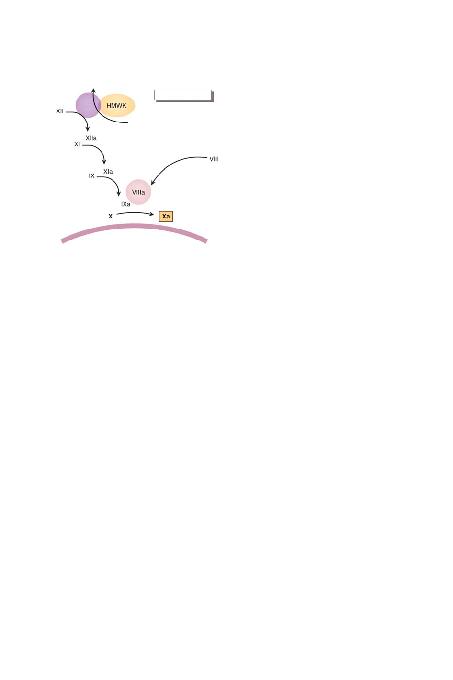
ve PK eksiklikleri klinik kanamaya yol açmazlar.
tor) aktif hale getirir. Faktör IXa faktör X'un faktör Xa olufl-
turacak flekilde bölünmesini sa¤lar, sonra da ortak yol bafl-
lar. Faktör IXa ile X aras>ndaki reaksiyon kalsiyum ve fos-
folipid varl>¤>nda ba¤lanma ile kolaylaflt>r>l>r; PTZ'de kulla-
n>lan tescilli etkinlefltirici ayraç da fosfolipid içerir. Fizyolo-
jik ortamda, fosfolipid yüzeyini trombositler sa¤lar (özellik-
le de etkin hale geldiklerinde). Faktör X'un faktör IXa tara-
f>ndan faktör Xa'ya dönüfltürüldü¤ü reaksiyonda maksium
h>z>n sa¤lanabilmesi için etkinleflmifl kofaktör VIII'nin bu-
lunmas> gerekir.
tirilirken, 2 ard>fl>k peptid ba¤> 2 zincirli bir serin proteaz
oluflturacak flekilde bölünür. Faktör VIII karaci¤erde, me-
gakaryositlerde ve vücudun çeflitli bölgelerindeki retikülo-
endotelial dokularda üretilir. Küçük bir molekül olan fak-
tör VIII dolafl>mda çok daha büyük bir protein olan vWF'e
ba¤l> halde bulunur. Bu iliflki son derece önemlidir. von
Willebrand hastal>¤> olan bireylerde vWF düzeyi azald>¤>
zaman (Bkz. 32. Bölüm) ba¤lanmam>fl faktör VIII plazma-
dan uzaklaflt>r>l>r ve düzeyi oldukça azal>r. Faktör X etkin-
lefltirilirken kofaktör olarak kat>l>m sa¤layabilmesi için fak-
a'ya dönüfltürülmesi gerekir.
li¤i) ve hemofili B (faktör IX eksikli¤i) ad>yla bilinen majör
kanama bozukluklar>na yol açar. Hemofili nadir bir bozukluk
(hemofili A 1:5,000 erkek do¤umu; hemofili B 1:30,000 er-
kek do¤umu) olmas>na ra¤men, bu hastal>¤>n cinsiyetle ba¤-
lant>l> ailevi bir kanama bozuklu¤u olarak tan>mlanmas> çok
eski ça¤lara dayan>r. Bu durum k>smen, a¤>r hemofili hastala-
r>nda do¤umdan hemen sonra bafllayan ve çocukluk ça¤>nda
da s>k aralarla tekrarlamaya devam eden majör kanamalar>n
görüldü¤ü gerçe¤ini yans>t>r. Bundan baflka, ailede hemofili
mutasyonu bir kez ortaya ç>kt>¤>nda, ekspresyonunda çok az
de¤ifliklik meydana gelerek nesiller boyunca sürdürülebilir.
Bununla birlikte, olgular>n üçte birinde hemofili yeni spontan
mutasyon (genellikle anneye ait gamette) olarak da ortaya ç>-
kabilir.
geni X kromozomu üzerinde bulunan 186 kb boyutunda çok
büyük bir gendir. Hemofili A hastalar>n>n yaklafl>k yar>s>nda
faktör VIII geninin büyük bir bölümünde (intron 22) inversi-
yon bulunur ve bu da faktör VIII üretiminin bozulmas> ile so-
nuçlan>r. Geri kalan hastalarda çeflitli nokta mutasyonlar>, in-
sersiyonlar ve X kromozom genomunun büyük k>s>mlar>n>n
delesyonu gibi faktör VIII antijen ve aktivite düzeyinin çok
düflmesine yol açan (genellikle normalin %1'inin alt>nda) de-
¤ifliklikler bulunur. Missens mutasyonlar da a¤>r hastal>¤a ne-
den olabilirler. Stop kodon, nokta mutasyonu ve minör deles-
yon gibi di¤er mutasyonlar genellikle faktör VIII düzeylerinin
%1'in üzerinde oldu¤u daha hafif bir hastal>¤a neden olurlar.
Baz> hastalarda ifllevsel aç>dan anormal bir protein üretilir; im-
munolojik faktör VIII antijen ölçümü (protein düzeyi normal-
dir) ile faktör VIII aktivitesini saptamaya yönelik koagülasyon
testi (aktivite düflüktür) aras>ndaki uyumsuzluk da bu duru-
mu yans>t>r.
aktivitesi %50-150 aras>ndad>r ve havuzlanm>fl normal plaz-
madaki düzey %100 aktiviteyi tan>mlar. A¤>r hemofili hasta-
lar>nda faktör VIII aktivite düzeyleri normalin %1'inin alt>n-
dad>r (<1 IU/mL) ve tan> s>k geliflen spontan eklem, kas ve ya-
flamsal organ kanamalar> nedeniyle genellikle çocukluk ça¤>n-
da koyulur. Bu hastalarda s>k s>k faktör VIII replasman> gere-
kir, fakat o zaman bile ilerleyici ve deforme edici artropati ris-
ki geliflme riski vard>r.
hastalarda cerrahi giriflim veya travma ile kanama riski hala
vard>r, fakat spontan hemartroz ve hematomlar konusunda
çok daha az zorluk yaflarlar. Faktör düzeyleri %6-30 aras>nda