evaluation.
and immunologic disease.
complement disorders.
n>mlanm>flt>r ve bunlar>n bir k>sm>nda genetik defektler tan>mlanm>fl-
t>r.
altta olabilecek nedenler ekarte edilmelidir.
li¤i; fagositer sistemle iliflkili immün yetmezlik ve kompleman sistem
anormallikleri olarak ayr> kategorilerde incelenir. Baz> T hücre iliflkili
immün anormallikler ayn> zamanda B hücre anormalliklerine de ne-
den olur. Bir çok primer immün yetmezlik çocukluk ça¤>nda görülse
de s>kl>kla B hücre immün yetmezlikleri geç eriflkin dönem gibi ileri
zamanlarda ortaya ç>kar. Hekim taraf>ndan karakteristik bulgular fark
edilmeli ve tedavi bafllanmal>d>r.
semptomlar>, bafllang>çta yap>lmas> gereken laboratuvar tekniklerini
ve tedavi yaklafl>mlar>n> tart>flmakt>r. Tüm primer immün yetmezlik-
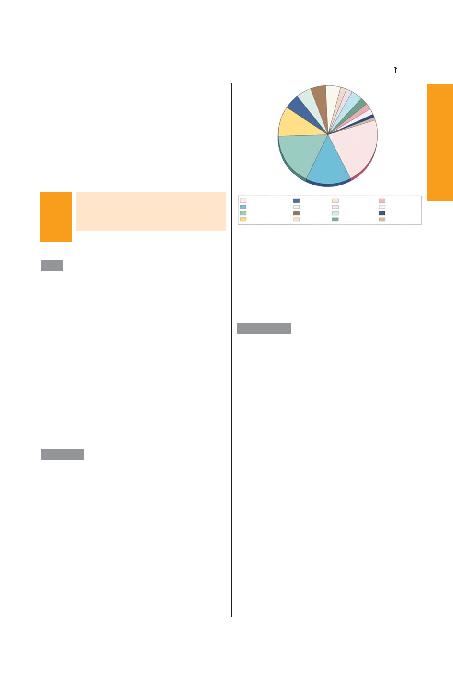
lerin yaklafl>k %65'ini antikor eksikleri, %15'ini kombine T ve B hücre
defektleri, %5'ini hücresel immün yetmezlikler, %10'unu fagositik
hücre anomalileri ve %5'inide kompleman eksikleri oluflturur (fiekil
271-1).
sesif, otozomal resesif veya otozomal dominant olarak tan>mlanm>flt>r.
Birçok immün yetmezlik T ve B hücrelerinin gelifliminde veya prekür-
sör hücrelerin immün hücrelere dönüflmesinde görevli genlerde olu-
flan mutasyonlarla geliflir. Birçok dokuda ortaya ç>kan di¤er genetik
anormallikler immün yetmezlikle birlikte komplike multisistemik has-
tal>klara yol açar. Ayn> immünolojik yollarla oluflan baz> genetik mu-
tasyonlar benzer klini¤e ve laboratuvar bulgular>na sahiptir.
na ve yap>s>na ba¤l> olarak farkl> klinik fenotipler oluflturabilirler. Ör-
ne¤in; rekombinasyon aktive edici gen (RAG)-1 ve RAG-2 genlerinde-
ki mutasyon a¤>r kombine immün yetmezlik hastal>¤>na ve Omenn
sendromuna neden olabilir. Wiskot-Aldrich sendromu, gen dizilimle-
rindeki mutasyon lokalizasyonlar>n>n çeflitlili¤ine di¤er bir örnektir.
Mutasyona u¤ram>fl bir proteinin ekspresyonu klinik olarak farkl> fe-
notiplerin ç>k>fl>na neden olur.
rekti¤i üzerinde durulmaktad>r. Baz> bakteriler ve viral patojenlerle
belirli yetmezliklerin iliflkisi oldu¤undan bu bilgi hekime ay>r>c> tan>
ve uygun laboratuvar tetkiklerin seçimi aç>s>ndan yard>mc> olur.
Sistemlerinden Elde Edilen
düflük virülansl> gram negatif bakterilerle oluflan tekrarlay>c> lenf ade-
nit ve apseler, fagosit fonksiyonlar>nda problem oldu¤unu düflündü-
rebilir. Staphylococcus epidermidis, Pseudomonas, Burkholderia cephacia gi-
bi beklenmeyen patojenlerle geliflen enfeksiyonlar, löksit fagasitoz bo-
zukluklar>n> düflündürebilir. Yine fagosit defektleri olan hastalarda
katalaz pozitif Staphylococcus aureus ile tekrarlayan deri enfeksiyonlar>
görülebilir. Bu enfeksiyonlar>n s>n>rland>r>lmas>nda efektif fagositoz
ve superokside ba¤l> öldürme rol al>r. Gecikmifl umbulikal kord düfl-
me hikâyesi ve kötü yara iyileflmesi lökosit adezyon defektlerini dü-
flündürür. Kronik granülomatöz hastal>kta, süpüratif lenf adenite rast-
lan>r. Ayr>ca enfekte dokulardan gram negatif bakteriler izole edilirse
bu hastal>¤a tan> koymak için iyi bir kan>t bulunmufl olur. Fagositik
hücrelerdeki immün yetmezlik sonucu mukus membranlar da etkile-
nir. Lökosit adezyon defekti gibi fagositik hücre defektlerinde oral ül-
serler görülür. C5, C9 gibi geç kompleman kompenent eksikliklerinde
Neiseria türleriyle oluflan enfeksiyonlara (Neiseria menengitidis menen-
jiti, Neiseria gonorrhoeae septik artiriti gibi) s>k rastlan>l>r. C3b, opsani-
zasyon ve fagositozun kolaylaflt>r>lmas>nda görevli oldu¤u için C3 ek-
sikli¤inde özellikle gram negatif bakterilerle a¤>r septisemiler görülür.
siyonlar görülür. Sinüzitin de¤erlendirilmesinde farinks ve burun bofl-
luklar>n muayenesi de önemlidir. Posterir farinkste kald>r>m tafl> man-
m>.
eksikli¤i vakalar>n yar>s>n> oluflturur. Di¤er eksiklikler infantlar>n geçici hipoga-
maglobülinemisi (THI), kronik granülomatöz hastal>k (CGD), antikor eksiklikleri
(Ab def), DiGeorge sendromu, Hiper IgM sendromu, di¤er T hücre defektleri, X'e
ba¤l> agamaglobülinemi (XLA), kompleman eksiklikleri, kombine defektler (def),
herediter anjioödem (HANE), fagositk anormallikler (ab), a¤>r kombine immün
eksiklik (SCID) ve nötropeniler.
THI
Di George
XLA
Kombine eksiklik